
 Newsletter ZPP
Newsletter ZPPWarszawa, 16 listopada 2023 r.
Komentarz ZPP w sprawie nowelizacji unijnego prawa farmaceutycznego
- 26 kwietnia 2023 r. Komisja Europejska przedstawiła projekty przepisów, za pomocą których zamierza zreformować unijne prawo farmaceutyczne.
- Na poziomie unijnym jest to największa nowelizacja prawa farmaceutycznego
od 20 lat. - Komisja Europejska stawia przed omawianymi regulacjami istotne cele, w zakresie:
- stworzenia wspólnego rynku oraz zapewnienia powszechnego i równego dostępu do tanich i skutecznych produktów leczniczych dla pacjentów zamieszkujących wszystkie kraje Unii Europejskiej;
- zaoferowania atrakcyjnego i sprzyjającego innowacjom systemu badań, rozwoju i produkcji leków w Europie;
- zmniejszenia obciążeń administracyjnych i znacznego przyspieszenia procedur wydawania pozwoleń na dopuszczenie do obrotu produktów leczniczych, tak aby mogły one jak najszybciej trafiać do pacjentów;
- zwiększenia dostępności i zapewnienia pacjentom stałych dostaw produktów leczniczych, bez względu na ich miejsce zamieszkania we Wspólnocie;
- zajęcia się kwestią oporności na środki przeciwdrobnoustrojowe w ramach unijnego podejścia „One Health”;
- ograniczenia wpływu produkcji i konsumpcji produktów leczniczych na środowisko naturalne
- 19 października 2023 r., podczas posiedzenia Komisji do Spraw Unii Europejskiej, podsekretarz stanu w MZ Maciej Miłkowski przedstawił stanowisko polskiego rządu ws. proponowanych zmian, w którym:
- aprobuje się kierunek zmian przyjęty przez Komisję Europejską i dostrzega wyzwania związane z brakiem dostępności leków oraz antybiotykoopornością;
- wskazuje się, że poszczególne rozwiązania proponowanego pakietu wymagają doprecyzowania – tak aby możliwe było osiągnięcie zakładanych przez Komisję Europejską celów;
- dostrzega się rozdźwięk interesów producentów leków innowacyjnych i generycznych;
- podkreśla się, że celem dyskutowanych przepisów jest zwiększenie dostępności leków dla pacjentów, a nie wsparcie danej grupy przedsiębiorców.
- 17 listopada 2023 r. Ministerstwo Zdrowia zaplanowało spotkanie Zespołu ds. rewizji unijnych przepisów lekowych, który – wraz z przedstawicielami interesariuszy branżowych i społecznych – ma dyskutować projektowane rozwiązania.
Ograniczona dostępność produktów leczniczych jest zauważalna nie tylko na rynku polskim, ale na całym rynku europejskim. Wszystkie kraje unijne mierzą się z problemem braku surowców, zwiększonymi kosztami produkcji, problemami logistycznymi oraz konkurencją ze strony rynków azjatyckich.
W związku z powyższym kluczowe jest podjęcie działań mających na celu dostosowanie obecnych regulacji farmaceutycznych – nie zmienianych istotnie przez ostatnie 20 lat – do dzisiejszych realiów, potrzeb i wyzwań. Konieczne jest zwiększenie atrakcyjności rynku europejskiego dla producentów i dystrybutorów w celu zachęcenia ich do przeniesienia produkcji do Unii Europejskiej.
Planowane rozwiązania – mimo szczytnych haseł uzasadniających ich wdrożenie – wydają się jednak niewystarczające. Koncentrują się na kilku obszarach (przede wszystkim na zmianach w procesie rejestracyjnym i okresie wyłączności rynkowej dla nowo rejestrowanych produktów), przerzucając obowiązki w zakresie zapewnienia dostępności na producentów. Oferowane zachęty mają charakter wirtualny (np. formalne skrócenie procesu rejestracji produktu do 180 dni lub teoretyczne wydłużenie okresu wyłączności rynkowej) i nie zawierają realnego pakietu wsparcia, ani zachęt do inwestycji dla przedsiębiorców działających na kluczowym dla gospodarki rynku.
Planowane przepisy pomijają jednocześnie kwestie, z którymi branża boryka się od lat, a które wymagałyby uwspólnienia na poziomie europejskim (np. problem wczesnego dostępu do terapii, wdrożenia nowoczesnego łańcucha dystrybucji, dostawy leków do domu pacjenta, presji cenowej ze strony płatnika publicznego).
Z uwagi na powyższe konieczna jest szczegółowa analiza planowanych regulacji pod kątem adresowania przez nie potrzeb nie tylko regulatora ale przede wszystkim pacjentów i przemysłu oraz dyskusja z regulatorem lokalnym a następnie europejskim w celu zaadresowania tychże potrzeb w projektowanych regulacjach.
26 kwietnia 2023 roku Komisja Europejska zaprezentowała projekty przepisów dzięki, którym planuje zreformować unijne prawo farmaceutyczne.
W związku z przyjęciem 26 kwietnia 2023 r. przez Komisję Europejską pakietu zmian europejskiego prawa farmaceutycznego i jednoczesnym rozpoczęciem procesu legislacyjnego w Radzie i Parlamencie Europejskim przedstawiamy stanowisko do projektu szczególnie z uwagi na fakt pojawiających się w przestrzeni publicznej tezy, jakoby przedmiotowe projekty ograniczą innowacyjność Europy względem innych rejonów świata. Uprzejmie informujemy, że nie zgadzamy się, że reforma prawodawstwa farmaceutycznego UE osłabi konkurencyjność przemysłu farmaceutycznego Unii Europejskiej (dalej UE). Szczególnie ma to znaczenie z uwagi na fakt, że termin na zgłaszanie poprawek w Komisji ENVI Parlamentu Europejskiego upłynął w połowie listopada 2023 r.
Spadek globalnej konkurencyjności europejskiego sektora farmaceutycznego w zakresie badań i rozwoju nie jest powiązany z erozją własności intelektualnej, ponieważ, od lat 90tych UE konsekwentnie zwiększała zachęty regulacyjne i monopole w zakresie własności intelektualnej. Każda nowa ochrona praw własności intelektualnej lub ochrona regulacyjna (patenty na produkty w ramach TRIPS, SPC, najdłuższe na świecie wyłączności regulacyjne i rynkowe, wyłączność na produkty sieroce, wyłączność pediatryczna i przedłużenie SPC) zostały wprowadzone w określonym celu, jakim jest uczynienie Europy światowym liderem w zakresie innowacji badawczo-rozwojowych.
Jednak to wzmocnienie zabezpieczeń monopolistycznych dokładnie odpowiada względnemu spadkowi badań i rozwoju w Europie w porównaniu z Chinami i USA. Pokazuje to, że twierdzenie, że „więcej monopolu prowadzi do większej liczby badań i rozwoju” jest fałszywe. Co gorsza, te środki monopolistyczne bezpośrednio przyczyniły się do przenoszenia produkcji leków poza Europę, chociaż pochwalamy wysiłki UE mające na celu skorygowanie tego za pomocą reform.
Natomiast środki polityczne, które pobudziły konkurencję ze strony sektora leków równoważnych (generycznych) w pełni spełniły pokładane w nich nadzieje. Przepisy dotyczące leków równoważnych (generycznych) pobudziły bardzo potrzebną konkurencję, podwoiły dostęp do leków w Europie i zmniejszyły presję na budżety opieki zdrowotnej. Zasady dla leków biologicznych równoważnych uczyniły Europę światowym liderem w tej technologii i przyczyniły się do znacznych inwestycji w produkcję leków biologicznych w UE.
Dlatego konieczne jest, aby strategia farmaceutyczna dla Europy w dalszym ciągu wspierała sektor leków równoważnych i biosimilarów gwarantujący bezpieczeństwo lekowe Europy.
W ocenie skutków prawodawstwa farmaceutycznego podkreślono (s. 43), że „trudno ustalić bezpośredni związek między zachętami UE a konkurencyjnością UE, ponieważ chociaż zachęty zwiększają atrakcyjność rynków UE, nie wpływają one na pochodzenie geograficzne leków. Około 20% nowych leków dopuszczonych do obrotu w UE pochodzi z UE, pozostałe pochodzą głównie z USA, Wielkiej Brytanii, Szwajcarii i Japonii, które w równym stopniu kwalifikują się do wszystkich zachęt UE. Podobnie innowacyjne przedsiębiorstwa z siedzibą w UE mogą skorzystać z zachęt gdzie indziej, jeśli będą tam sprzedawać swoje produkty.
W czerwcu 2016 r. Rada zwróciła się do Komisji o przeprowadzenie opartej na dowodach analizy wpływu mechanizmów zachęt, zwłaszcza SPC. Zlecono dwa badania. Jeden z Instytutu Maxa Plancka [1] kwestionuje, czy dostępność ochrony patentowej lub SPC wpływa na decyzje przedsiębiorstw dotyczące lokalizacji obiektów badawczych w tej czy innej jurysdykcji, podkreślając, że inne czynniki mają prawdopodobnie większe znaczenie.
W drugim badaniu Copenhagen Economics [2]argumentowano, że SPC mogą odegrać rolę w przyciąganiu innowacji do Europy, wskazując, że podatki, edukacja i inne czynniki są prawdopodobnie ważniejsze w tym względzie.”
Okresy wyłączności regulacyjnej i vouchera na wyłączność
Pakiet farmaceutyczny zmienia zasady przyznawania firmom tzw. wyłączności regulacyjnej, tzn. czasowej ochrony podmiotów, które wprowadziły dany lek jako pierwszy, przed konkurencją rynkową. Skutkiem zmian ma być wydłużenie maksymalnej ochrony – i tak już dłuższej niż np. w Stanach Zjednoczonych – z obecnych 11 lat do 13 lat.

Tymczasem brak jest dowodów na korelację pomiędzy wydłużaniem ochrony opartej na wyłączności a poziomem innowacyjności – dane na temat kierunków importu do Unii Europejskiej innowacyjnych leków (USA, Szwajcaria, Wielka Brytania, Japonia) świadczą o czymś przeciwnym.
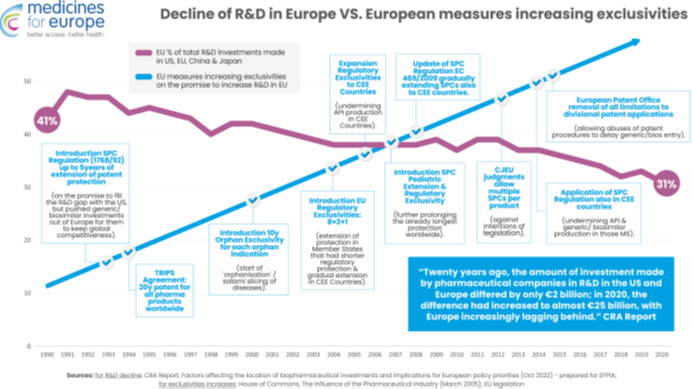
Ponadto proponowane nowe zasady przyznawania ochrony są tak niejasne, że wprowadzają niepewność co do tego, kiedy w konkretnym przypadku możliwa będzie sprzedaż leku konkurencyjnego. Nie można zapominać, że przygotowanie produktu generycznego wymaga przeprowadzenia prac badawczo-rozwojowych i badań klinicznych (badań biorównoważności) oraz przygotowania dokumentacji rejestracyjnej. Wytwórca produktów generycznych musi z kilkuletnim wyprzedzeniem wiedzieć, kiedy będzie mógł złożyć wniosek o wydanie pozwolenia na dopuszczenie do obrotu, tak by odpowiednio rozplanować w czasie prowadzone prace.
W przypadku, gdy w toku tych prac nagle okaże się, że wyłączność danych została przedłużona, może się okazać, że część przeprowadzonych badań czy przygotowanych dokumentów, które spełniały wymagania przewidziane prawem w dniu, w którym upływał sześcioletni okres wyłączności danych, nie będzie ich spełniać w dniu, w którym upłynął przedłużony okres wyłączności danych. Ponadto w czasie trwania wyłączności danych w UE, producenci generyków mogą rejestrować produkty lecznicze za granicą i tam je zbywać. Jednakże w wielu krajach warunkiem uzyskania dopuszczenia do obrotu jest posiadanie pozwolenia na dopuszczenie do obrotu w państwie wytwórcy. Tym samym wydłużanie wyłączności danych zmniejszy konkurencyjność producentów z Unii Europejskiej w stosunku do producentów z Indii, Chin, czy USA. Tam nie obowiązują bowiem tak długie terminy wyłączności danych, jak w Unii Europejskiej. Dlatego też ewentualne dalsze dodatkowe okresy ochronne powinny być przyznawane wyłącznie w zakresie wyłączności rynkowej, a nie wyłączności danych.
Dodatkowo pakiet farmaceutyczny przewiduje instytucję przenoszalnego vouchera na wyłączność (TEV, Transferrable Exclusivity Voucher). Firma wprowadzająca na rynek nowy lek przeciwbakteryjny otrzyma prawo 12-miesięcznej wyłączności, które może wykorzystać dla innego leku lub sprzedać osobie trzeciej. Zaproponowana konstrukcja, choć miałaby zachęcać do prac badawczo-rozwojowych nad nowymi antybiotykami, w rzeczywistości może otworzyć przestrzeń dla nadużyć i antykonkurencyjnych zachowań. Warto rozważyć inne metody wsparcia rozwoju antybiotyków jakie zostały zaproponowane chociażby w Szwecji czy w Wielkiej Brytanii tj. roczny program gwarancji przychodów wydaje się skutecznym narzędziem, które może zachęcić twórców antybiotyków do inwestowania w badania i rozwój poprzez zapewnienie gwarantowanych przychodów. Modele szwedzkie i brytyjskie pokazują, że wdrożenie gwarancji dochodów jest praktycznym i wykonalnym rozwiązaniem, które zostało przetestowane i przyniosło pozytywne rezultaty.
Nawet europejskie stowarzyszenie leków monopolistycznych EFPIA nie wskazuje kwestii własności intelektualnej (IP) jako koniecznych do relokacji produkcji.

Każdy rok opóźnienia konkurencji to wymierne straty dla pacjentów i narodowych płatników, a jeden rok to strata rzędu przynajmniej setek milionów euro.

Mając na uwadze powyższe apelujemy o:
– ustanowienie maksymalnego jednolitego okresu wyłączności danych wynoszącego 6 lat oraz wyłączności rynkowej w wymiarze podstawowym 2 lat, przy czym wyłączność rynkowa mogłaby być rozszerzana o dodatkowe okresy stanowiące zachęty obecnie przewidziane w projekcie dyrektywy, niemniej jednak łączny czas trwania ochrony rejestracyjnej nie powinien być dłuższy niż obecnie obowiązujące okresy, tj. maksymalnie 11 lat;
– o wyraźne wskazanie w przepisach, w przypadku, gdy wydłużenie wyłączności danych albo wyłączności rynkowej miałoby być zależne od zapewnienia dostępności produktu leczniczego na rynku dla pacjentów, że taka dostępność oznacza nie tylko wprowadzenie produktu leczniczego na rynek i zapewnienie dostaw pokrywających zapotrzebowanie pacjentów, ale także uzyskanie refundacji dla tego produktu leczniczego, ewentualnie dostaw produktu leczniczego na rynek UE lub wycofanie wniosku o refundację w którymkolwiek z państw członkowskich powinno skutkować odebraniem przyznanej wyłączności;
- – rezygnację z instytucji TEV i zastąpienie jej schematem gwarantowanego rocznego przychodu (Annual Revenue Guarantee Scheme – wypracowanym przez EU-Joint Action on Antimicrobial Resistance and Healthcare-Associated Infections, EU-JAMRAI), który wynagradza innowacje, jednocześnie promując odpowiedzialną konsumpcję antybiotyków.
Zmiany w zakresie ochrony środowiska
Popierając kierunkowe wszelkie działania zmierzające do poprawy stanu środowiska naturalnego należy podkreślić, że już obecnie europejski przemysł farmaceutyczny musi spełnić najwyższe standardy środowiskowe. Paza zmianami w pakiecie farmaceutycznym, obecnie toczą się zaawansowane prace legislacyjne w Parlamencie Europejskim i Radzie w zakresie chociażby ścieków komunalnych, które wpłyną w istotny sposób na europejski przemysł farmaceutyczny produkujący leki równoważne (generyczne i biologiczne równoważne) co przełoży się na wzrost kosztów wytwarzania tych leków. Mając to na uwadze należy zważyć również perspektywę aby słuszne cele i założenia nie stały się barierą konkurencyjną i nie doprowadziły do wyeliminowania tej części branży farmaceutycznej
z Europy. Szczególnie ma to znaczenie z uwagi na braki leków podstawowych w Europie
i konkurencję azjatycką która z jednej strony jest dotowana finansowo przez swoje rządy,
a z drugiej nie podlega tak rygorystycznym przepisom.
Wpływ na konkurencyjność i MŚP
W przypadku małych i średnich przedsiębiorstw w ocenie skutków stwierdzono:
- (s. 60) „Jeśli chodzi o wpływ na konkurencyjność, proponowane zachęty nie uwzględniają rozróżnienia geograficznego, w równym stopniu zapewniają ochronę regulacyjną produktów opracowanych w UE lub gdziekolwiek na świecie, co zapewnia równe warunki działania dla przedsiębiorstw z siedzibą w UE i z krajów trzecich. Chociaż ramy regulacyjne UE są atrakcyjne dla R&D, konkurencyjność zależy również od wielu innych czynników, m.in. system podatkowy i zachęt; dostępne dotacje, pożyczki i inne źródła finansowania (np. Akcelerator Europejskiej Rady ds. Innowacji); pula talentów; bliskość najlepszych środowisk akademickich; infrastruktury do badań klinicznych; wielkość rynku; bezpieczeństwo łańcuchów dostaw; korzystne decyzje refundacyjne.”
- (strona 61) „Podobnie zachęty dla UMN przyniosłyby korzyść MŚP, które na ogół są skłonne dokonywać inwestycji na wczesnym etapie w obszarach wysokiego ryzyka, zwiększając wartość ich aktywów, nawet jeśli zostaną przejęte przez dużą firmę farmaceutyczną na późnym etapie rozwoju. MŚP korzystają już ze zwolnień i obniżek z opłat za procedury regulacyjne, a dzięki nowym środkom horyzontalnym MŚP będą korzystać ze zoptymalizowanego wsparcia naukowego z większym prawdopodobieństwem pomyślnego uzyskania zezwolenia. Ogólnie rzecz biorąc, wraz ze wzrostem inwestycji w badania i rozwój w dziedzinie biofarmaceutyki oraz rosnącym udziałem MŚP wśród R&D, MŚP z branży biofarmaceutycznej w UE i poza nią miałyby doskonałe perspektywy na przyszłość.”
Komisja Europejska z jednej strony mówi o skrócenie okresów wyłączności leków, ale z drugiej, zaproponowane przepisy de facto otwierają cały wachlarz możliwości ich przedłużania. Maksymalny okres wyłączności na rynku dla leku – oczywiście poza 20 letnią ochroną patentową – według nowej projektowanej dyrektywy i rozporządzenia ma wynosić 13 lat,
a dziś jest to 11 lat.
Ponadto Komisja Europejska zaproponowała również tzw. pakiet IP – obecnie równolegle procedowanym w Radzie i Parlamencie Europejskim, w którym firmy monopolistyczne uzyskują możliwość wprowadzenia jedną europejską procedurą, SPC we wszystkich krajach UE. Dziś muszą ubiegać się o to państwo po państwie. I mimo że w pakiecie farmaceutycznym wprowadzane są regulacje skracające procedury rejestracyjne, wprowadzony na początku lat 90. XX w.
SPC mający rekompensować długość procesu rejestracji – nie zostaje skrócony.
Poza tym jest wiele projektów, które są dedykowane dla rozwoju badań nad nowymi lekami, jak chociażby wsparcie ich unijnymi funduszami – mechanizmy IPCEI czy inicjatywami publiczno – prywatnymi największymi na świecie – Innovative Medicines Initiative,
a teraz Innovative Health Initiative, w których połowa funduszy pochodzi od wszystkich obywateli UE, a połowa z firm monopolistycznych reprezentowanych przez COCIR, EFPIA / Vaccines Europe, EuropaBio, MedTech Europe, a budżet całkowity budżet IHI na lata 2021–2027 wynosi 2,4 miliarda euro.
To wszystko – mimo krytycznych głosów ze strony firm monopolistycznych- wskazuje na otwartość i wsparcie UE w tym zakresie. Warto, aby wsparcie otrzymywał też przemysł produkujący leki konkurujące na rynku ceną, bo to właśnie ich brakuje w aptekach. A w tym obszarze fundusze unijne nie są już tak szczodre.
Z pewnością nie należy okresu monopolu przedłużać, bo europejscy pacjenci dłużej będą czekać na pojawienie się konkurencji cenowej na rynku leków, a płatnicy narodowi, w tym nasz NFZ, zmuszeni zostaną do ponoszenia wyższych kosztów refundacyjnych. Każdy dzień opóźnienie konkurencji to wielomilionowe starty dla budżetów ochrony zdrowia.
Zobacz: 16.11.2023 Komentarz ZPP w sprawie nowelizacji unijnego prawa farmaceutycznego












{kind=link}